6.4. Синдром диссеминированного внутрисосудистого свертывания
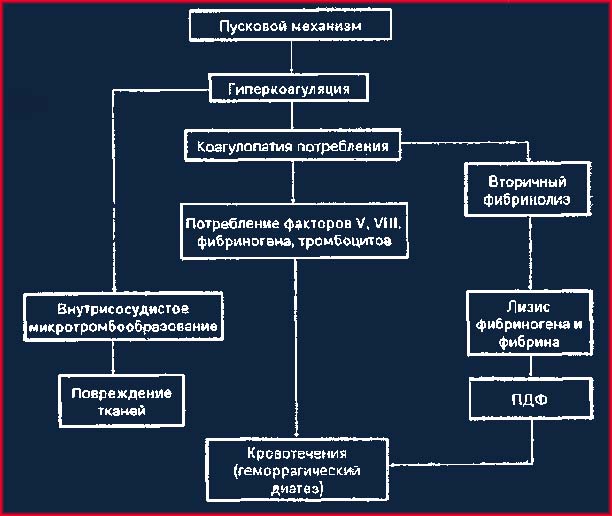
ДВС-синдром, известный также как тромбогеморрагический, или коагулопатия потребления, осложняет патологические процессы и состояния, например массивную кровопотерю, травматические повреждения, различные виды шоковых состояний, злокачественные новообразования, тяжелые деструктивные процессы в органах, массивные гемотрансфузии, гипоксические состояния, отравления ядами и др. [Балуда В. П., 1977]. В основе синдрома лежит рассеянное свертывание крови в мелких сосудах, блокирующее их и вызывающее глубокие нарушения функции органов. Развиваются тяжелая гипоксия и ацидоз, которые нередко приводят к гибели больного [Raby С., 1974]. В результате массивного коагуляционного процесса, истощающего коагуляционный потенциал организма, возникает дефицит ряда факторов свертывания, ведущий затем к неконтролируемым профузным кровотечениям (схема 6.4).
Схема 6.4. Развитие ДВС-синдрома
В основе многообразных и многочисленных пусковых механизмов ДВС-синдрома лежат три главных момента [Cash J. D., 1977]:
1) стимуляция коагуляционного процесса путем высвобождения тканевых факторов в кровоток. Это может иметь место, в частности, при обширных хирургических операциях, эпизодах внутрисосудистого гемолиза, диссеминации злокачественно перерожденной ткани;
2) активация тромбоцитарной агрегации, которая может возникнуть при септицемии, вирусной инвазии, болезни иммунных комплексов, появлении в циркуляции тромбина, образующегося в результате активации коагуляционных факторов;
3) тяжелые поражения эндотелия сосудистых стенок, возникающие в результате распространенного васкулита, при ожогах, ацидозе, инфекционном процессе, продолжительной гипотензии или гипоксии и активации как внешнего, так и внутреннего механизма коагуляции крови.
Следует подчеркнуть, что более чем в 60% случаев ДВС-синдром связан с сепсисом, вызываемым главным образом грам-отрицательной флорой. Нередко ДВС-синдром бывает обусловлен вирусной инфекцией, патогенной грибковой флорой, туберкулезными микобактериями.
В циркулирующей крови в результате ДВС-синдрома снижается содержание тромбоцитов, накапливаются продукты про-теолиза, которые оказывают антикоагулирующее и токсическое действие, усиливается процесс фибринолиза. Развивающийся тяжелый геморрагический синдром в ряде случаев может быть единственным клиническим проявлением этого сложного патологического процесса. Разнообразны провоцирующие ДВС-синдром патологические факторы. Это прежде всего образующиеся тканевые тромбопластины, коллаген, бактериальные коагулазы, воздействие ядов змей, образование иммунных комплексов. Различия названных факторов определяют клинико-патогенетическое разнообразие проявлений ДВС-синдрома. Однако конечный механизм развития синдрома всегда связан с образованием в системе микроциркуляции рыхлых масс фибрина и агрегатов клеток крови [Баркаган 3. С. и др., 1979; Балу-Да В. П., 1979; Баркаган 3. С., 1980, и др.].
С точки зрения патофизиологии образование сгустков на уровне капилляров обусловлено прежде всего избытком тромбина, под влиянием которого происходит активная трансформация фибриногена в фибрин.
В развитии ДВС-синдрома различают несколько клинических стадий. Каждая из них имеет специфическую клинико-лабораторную характеристику [Мачабели М. С., 1970; Баркаган 3. С., 1980],.
I стадия — гиперкоагуляция крови и образование внутрисо-судистых агрегаций клеток, формирование блокады микроциркуляции в органах (легкие, почки, печень и др.), комбинация, гиперкоагуляции с началом истощения свертывающих и противосвертывающих механизмов. Как правило, I стадия синдрома совпадает с острой фазой критического состояния.
II стадия — глубокое истощение факторов свертывания крови, развивающееся в результате потребления тромбоцитов, фибриногена, факторов V, VIII, XIII. В крови накапливаются ингибиторы свертывания крови и агрегации, а также продукты деградации фибриногена и фибрина, которые сами по себе обладают антикоагулянтной активностью. Возникают профузные кровотечения. Эта стадия характеризуется также максимальным истощением противосвертывающих механизмов, в частности антитромбина-Ш, который расходуется на инактивацию тромбина и ряда активированных протеолитических факторов свертывания. Снижается также концентрация плазминогена, который под влиянием активаторов трансформируется в плазмин.
III стадия — исходы и остаточные явления ДВС-синдрома (тромбозы и дистрофии органов).
Этиологическое множество форм ДВС-синдрома в значительной степени усложняет его диагностику. Однако преодоление трудностей облегчается тем, что при многих формах патологии ДВС-синдром является, как считает 3. С. Баркаган (1980), либо единственно возможным нарушением гемостаза, либо обязательным компонентом заболевания. В связи с этим клиническая диагностика ДВС-синдрома в значительной степени становится ситуационной, т. е. диагностика конкретного патологического состояния (сепсиса, кровопотери, распространенного ракового поражения, ожога) уже предполагает наличие ДВС-синдрома. Важным, хотя и не обязательным компонентом может быть наличие у больного клинически диагностируемых тромбогеморрагических явлений.
В хирургической практике связь тромбогеморрагических явлений с возникающим или развившимся ДВС-синдромом обусловлена значительной кровопотерей, массивной гемотрансфузией, тяжелой травмой, сепсисом, трансплантацией органов, искусственным кровообращением, ожогами, гипоксией и постгипоксическим синдромом, другими критическими состояниями. ДВС-синдром часто встречается в акушерской практике [Herbert W., Cefalo R. С., 1984; Sher G., Satland В. Е., 1985].
Хотя основы изучения расстройств микроциркуляции у больных в критических состояниях заложены в конце 40-х — начале 50-х годов [Knisely Н., 1947; Bloch A. S., Elliott S. Н., 1962, и ДР-], разработка учения о ДВС-синдроме является достижением последних лет. Вместе с тем опыт лечения ДВС-синдрома пока невелик во всем мире. Вполне возможно, что потребуется частично или полностью пересмотреть, а может быть, дополнить ряд положений. С нашей точки зрения, одно из таких положений относится к гипердиагностике ДВС-синдрома. В последние годы достаточно типичную картину ДВС-синдрома, подтвержденного лабораторными данными и динамикой их в процессе лечения, мы наблюдали лишь у 20 из 1000 больных, леченных по поводу различных критических Состояний.
Другой важный момент связан с определением роли, которую в происхождении ДВС-синдрома играют ятрогенные факторы, такие как массивная гемотрансфузия, гиперосмоляльный синдром, инфузия кровезаменителей и плазмоэкспандеров. Этот вопрос требует дополнительных исследований.
В настоящее время в литературе отсутствуют четкая информация и данные о связи ДВС-синдрома с гипоксией или постгипоксическим (с возможным участием гиперкапнии) состоянием. В нашей клинической практике получены доказательства подобной связи, которые обсуждены ниже.
С клинической точки зрения при диагностике ДВС-синдрома важно обнаружить следующие типичные симптомы: тромбоцитопению, снижение содержания фибриногена в плазме, повышение содержания ПДФ в плазме, наличие в мазке крови обломков эритроцитов (феномен фрагментации), положительные этаноловый и протаминсульфатный тесты (у некоторых больных), снижение концентрации факторов V, VII, VIII, IX, X и антитромбина-III, иногда снижение активности фактора XIII. Геморрагический компонент синдрома формируется главным образом в стадии гипокоагуляции (см. схему 6.4) и представляет собой опасную, хотя и не обязательную фазу ДВС-синдрома. Он проявляется кровотечением из всех мест повреждения тканей и особенно опасен при хирургических и акушерских ситуациях прежде; всего в силу обширности области геморрагии [McKay D. G., 1983]. Возникшее вначале в определенной области кровотечение становится генерализованным и дополняется неспровоцированными носовыми геморрагиями, кровотечениями в желудочно-кишечный тракт, забрюшинными гематомами, кровоизлияниями в различные органы и кровотечениями в плевральную и брюшную полости. Характерны неостанавливаемые кровотечения из всех мест проколов кожи.
Вытекающая из ран или из матки кровь не образует полноценных свертков либо часами не свертывается. Эти нарушения, обусловленные частично дефицитом фибриногена, возникают главным образом в результате активации фибринолиза и протеолиза, а также связаны с накоплением в плазме ПДФ, накоплением несвертывающихся или плохо свертывающихся фибри-ногеновых или фибрин-мономерных комплексов. Однако практически никогда не наблюдается полного исчезновения фибриногена из крови. По современным представлениям о патогенезе ДВС-синдрома, его развитие не связано исключительно с дефицитом фибриногена или с его патологией. Более того, известны случаи, когда этот синдром возникал при нормальном или даже повышенном уровне фибриногена.
В настоящее время убедительно доказано, что ДВС-синдром — комплексное заболевание, в основе которого лежит не только блокада микроциркуляторного сосудистого русла осаждающимся фибрином, агрегатами клеток и продуктами фибринолиза и протеолиза, но и повреждение стенок сосудов указанными элементами. Развивающийся сладж-синдром и ацидоз также вызывают дистрофию стенок сосудов. Важнейшим компонентом этого процесса является также патология тромбоцитов, которая служит одной из главных причин неостанавливаемых кровотечений при ДВС-синдроме [Kowalski D. S., SolumM., 1973].
Нарушения микроциркуляции в наибольшей степени выражены в легких и почках. В легких развивается картина, характерная для СДРВ с интерстициальным отеком легких, синдромом внутрилегочного шунтирования и нарушением диффузии газов через альвеолярно-капиллярную мембрану. Появляется и прогрессирует гипоксия, которая клинически проявляется одышкой, цианозом, снижением PaО2. Нарастает острая почечная недостаточность, которая выражается в олигурии, иногда анурии, низкой плотности мочи, появлении в моче белка и свободного гемоглобина, цилиндров, эритроцитов. Повышается содержание мочевины и креатинина крови.
При геморрагических проявлениях диагноз ДВС-синдрома можно ставить с уверенностью, если концентрация фибриногена в крови 1 г/л или менее, протромбиновый индекс менее 45%, а число тромбоцитов снижается до 90-109/л [Kunz F. et al., 1974]. Вследствие активации коагуляционной системы и нарастающего внутрисосудистого тромбоза развивается также вторичное усиление фибринолитической активности, которая ведет к образованию плазмина, растворяющего свертки фибрина и разрушающего циркулирующий фибриноген.
В плазме появляется большое количество ПДФ [Серов В.Н., Макацария А.Д., 1987], уровень которых при острых вариантах ДВС-синдрома превышает 80—100 мкг/мл. ПДФ можно определить экспресс-методом, используя специальные коммерческие наборы. У некоторых больных уровень ПДФ иногда не соответствует выраженности синдрома и остается невысоким. Это связано с угнетением фибринолитической системы и прогностически оценивается как неблагоприятный симптом, поскольку свидетельствует о необратимости блокады микрососудистого русла.
Удлинение тромбинового времени обычно связано с истощением коагулируемого фибриногена, увеличением концентрации ПДФ, которые в подобных случаях действуют как патологические антикоагулянты, а также с повышением уровня циркулирующего гепарина. Все это значительно увеличивает антикоагулянтный потенциал крови и обусловливает массивные кровотечения при низком уровне фибриногена. При увеличении тромбинового времени вдвое по сравнению с нормой кровотечение становится угрожающим.
Важное значение в диагностике ДВС-синдрома имеет выявление дефицита в плазме антитромбина-III, на долю которого приходится до 80% антикоагулянтного потенциала плазмы. Установлено, что дефицит антитромбина-III ниже 30% не совместим с жизнью, поскольку приводит к множественным тромбозам [Баркаган 3. С. и др., 1980] и не корригируется гепари-нотерапией. Развивающаяся в критических состояниях активация всех ферментных систем организма, включая фибринолити-ческую, комплементарную, калликреиновую, резко уменьшает в плазме содержание антитромбина-III, являющегося основным ингибитором активированных факторов и других протеаз. Происходит интенсивное потребление антитромбина-III на инактивацию всех протеаз, участвующих в процессе гемокоагуляции,— активированных факторов XII, XI, IX, X, II и др. Истощение содержания антитромбина-III существенно снижает антикоагулянтный и антитромботический эффект эндогенного и экзогенного гепарина.
Как мы уже указывали, ДВС-синдром имеет многофакторный патогенез, который еще не изучен до конца. В табл. 6.2 представлен ряд состояний, которые могут быть непосредственной причиной различных коагулопатий, в том числе ДВС-синдрома. Среди них есть указание на гипоксию и постгипоксический синдром. На возможную связь коагулопатий с хроническим гипоксическим состоянием, обусловленным врожденным пороком сердца, указывали Е.П. Степанян, А.И. Лагутина (1959) и Л.М. Терентьева (1964). Однако вероятность развития ДВС-синдрома при острой гипоксии в литературе практически не обсуждается. Между тем возможность и даже обязательность освобождения значительного количества тромбопластина и других тканевых коагулаз при гипоксическом повреждении клетки неоспорима. Это предполагает активацию коагуляционного потенциала, которую при гипоксии нередко можно подтвердить документально. В этой связи считаем важным сослаться на собственное наблюдение, подтверждающее связь ДВС-синдрома с гипоксией и гиперкапнией.
Больной X., 61 года, долгие годы страдавший бронхиальной астмой, поступил в отделение реанимации в связи с развитием астматического статуса. Опуская обстоятельства, предшествовавшие возникновению статуса, его причины, динамику развития и ряд обстоятельств лечения, укажем лишь, что прогрессирование статуса привело в конце концов к необходимости перевода больного на ИВЛ. Несмотря на многократную фибробронхоскопию, лаваж, массивную бронхолитическую терапию постепенно развились гиперкапния (РаСО2 65 мм Рт. Ст.) и гипоксия (Рао2 52 мм рт. ст.).
Максимально допустимый в создавшихся условиях режим вентиляции не улучшил состояние больного, и через 1 сут после начала ИВЛ стала нарастать гиперкапническая и гипоксическая кома. Бронхиальная блокада в этом периоде достигала высоких степеней (Рсо2 превышало 100 мм рт. ст.),. хотя выраженность гипоксии несколько снизилась: при FiO2 0,5 удавалось удерживать Рао2 на уровне 82 мм рт. ст. ИВЛ продолжалась 2 сут, после чего стал определяться гипокоагуляционный синдром: появились неостанавливаемые кровотечения из мест уколов на коже, постинъекционные гематомы под кожей. В отсутствие признаков внутреннего кровотечения стала нарастать анемия, которая не могла быть объяснена внешней кровопотерей: концентрация гемоглобина снизилась с 157 до 110 г/л. Исследование коагулологических показателей свидетельствовало о развитии ДВС-синдрома: количество тромбоцитов 70-109/л, АЧТВ 60с, содержание фибриногена 0,9 г/л, ПДФ 80 мкг/мл.
Развитие ДВС-синдрома можно было связать лишь с крайне тяжелым общим состоянием, обусловленным гипоксией, гиперкапнией и комой. Назначено лечение свежезамороженной плазмой (600 мл/сут) в сочетании с ин-фузией гепарина (500 ЕД/ч) через инфузомат. Явления ДВС-синдрома купированы через 1 сут. Улучшились также показатели газообмена. Однако нарастала тяжесть клинической симптоматики постгипоксического и постгиперкапнического поражения мозга, прогрессировала кома. Больной умер на 21-е сутки после начала астматического статуса при явлениях апалли-ческого синдрома.
Лечение. Лечение острого ДВС-синдрома представляет собой весьма трудную задачу прежде всего из-за обилия одномоментно действующих факторов. Прежде всего, если возможно, необходимо ликвидировать пусковой механизм синдрома. Для этого, например, устраняют гиповолемию, антибиотики вводят в соответственно повышенных дозах и только внутривенно,, пытаются при шоке устранить факторы, способствующие сосудистому стазу и высокой внутрисосудистой агрегации, корригируют ацидоз и добиваются адекватной оксигенации крови.
Главной задачей лечения ДВС-синдрома является прекращение процесса спонтанного внутрисосудистого свертывания крови. Наиболее быстро и эффективно это может быть достигнуто сочетанным введением главных ингибиторов тромбина — гепарина и антитромбина-III [Баркаган 3. С., 1976; Matsuda Tamotsu, 1988]. Гепарин вводят внутривенно капельно в дозе 2500—5000 ЕД в изотоническом растворе хлорида натрия, в растворе сухой плазмы, в нативной или свежезамороженной плазме в течение 3—4 ч, повторяя введение в течение суток с таким расчетом, чтобы суточная доза не превысила 10000—12000 ЕД. N. Cosson и соавт. (1976) рекомендуют вводить гепарин медленно капельным способом, чтобы суточная доза составляла 30—50 ЕД/кг. Именно в малых дозах гепарин дает наиболее выраженный эффект, направленный на ингибицию фактора Ха и тромбина. Введение гепарина сочетают с капельной инфузией свежезамороженной плазмы. Она является донатором антитромбина-Ш, которого в ней содержится 200—250% средней нормы, а также донатором плазминогена.
Если имеет место геморрагический диатез, необходимо осуществить адекватную заместительную терапию тромбоцитной массой, свежезамороженной плазмой или криопреципитатом, который также содержит достаточный запас фибриногена. Целесообразно вводить белковые препараты, например раствор протеина, 10 или 5% раствор альбумина. Плазмоэкспандеры при лечении ДВС-синдрома не показаны, так как способствуют поддержанию кровотечения. Не показаны также концентраты, содержащие фактор IX и фибриноген, или чистый препарат фибриногена, так как их переливание увеличивает массу внутрисосудистого фибрина и, следовательно, усиливает внутрисосудистый тромбоз.
Для оценки эффективности лечения, а также для определения направления дальнейшего лечения необходимы повторные многократные исследования коагуляционного потенциала. Наиболее информативными показателями остаются тромбиновое время, число тромбоцитов в крови, содержание фибриногена в плазме, наличие ПДФ, протромбиновое время (индекс) и показатели фибринолитической активности.
Отношение к гепарину как одному из средств сочетанного лечения ДВС-синдрома, по литературным данным, неоднозначно. Имеются многочисленные данные, свидетельствующие как о его пользе, так и о его опасности при выраженном кровотечении, обусловленном ДВС-синдромом. Мы являемся сторонниками применения гепарина, однако при абсолютном условии остановки хирургического (акушерского) кровотечения. Как уже указывалось, предпочтительны малые дозы (не более 500 ЕД/ч) при одновременном введении свежезамороженной плазмы, содержащей антитромбин-Ш в высоких концентрациях. S. J. Ma-chin (1983) считает возможным увеличить дозу, если клиническое улучшение не наступило, но лабораторные показатели остаются стабильными. Относительная неэффективность гепаринотерапии в ряде случаев может быть связана с дефицитом антитромбина-III, что свидетельствует о необходимости комбинированного применения этих препаратов.
|
● |
● |
● |
● |
● |
● |
● |
● |